Industry Solutions
DP3® Platform
Why Corista
Resources
About Us
Contact Us
Industry Solutions
Healthcare
Increase efficiency and collaboration with an image management solution built by pathologists for pathologists.
Life Sciences
Conduct more studies and experiments with collaborative access to data and images across labs and locations.
DP3 Platform
DP3 Image Management
Discover how DP3 helps your organization become more efficient and productive.
Integrations
Explore DP3's seamless integrations to your favorite tools and applications.
Resources
Blog
Keep up with industry trends and get insights from our team of renowned scientists and pathologists.
Content Library
Explore best practices, watch webinars, and read how organizations drive efficiency with Corista.
News & Events
Learn what’s new with Corista and find out about upcoming events.
Corista Blog
Subscribe to the Blog
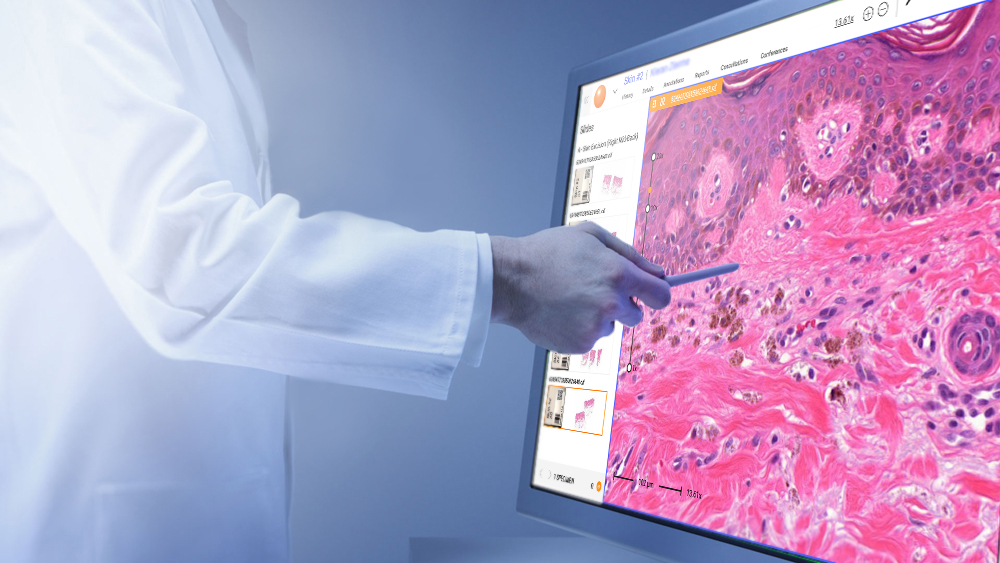
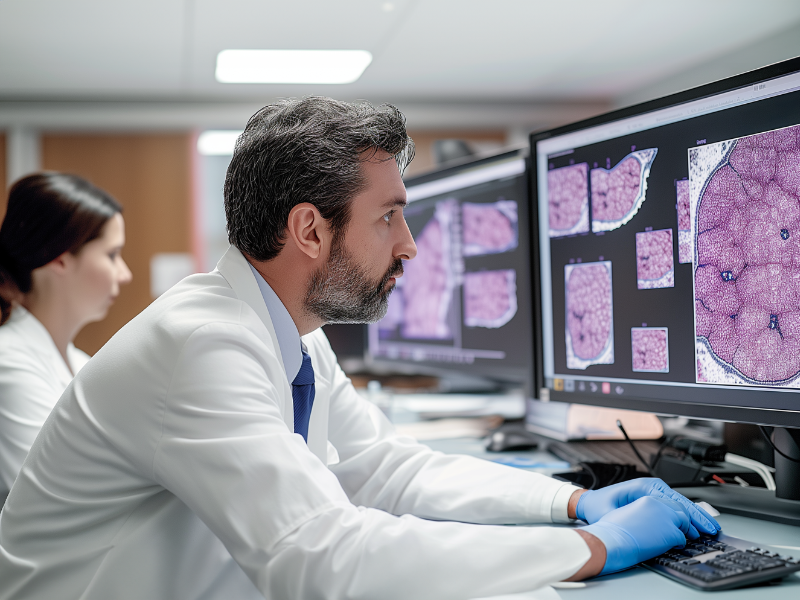
Digital Pathology
Pathology
Slide Management
Healthcare
Management
Studies/Reports
Challenges and Pitfalls Navigating Interoperability in Digital Pathology
Read Now
Connecting the Pieces: Why Interoperability Is Defining the Future of ...
Read Now
Pathology’s Digital Revolution: A Practical Guide to Systems Integration
Read Now
Transforming Clinical Workflow: Digital Pathology in Research and ...
Read Now
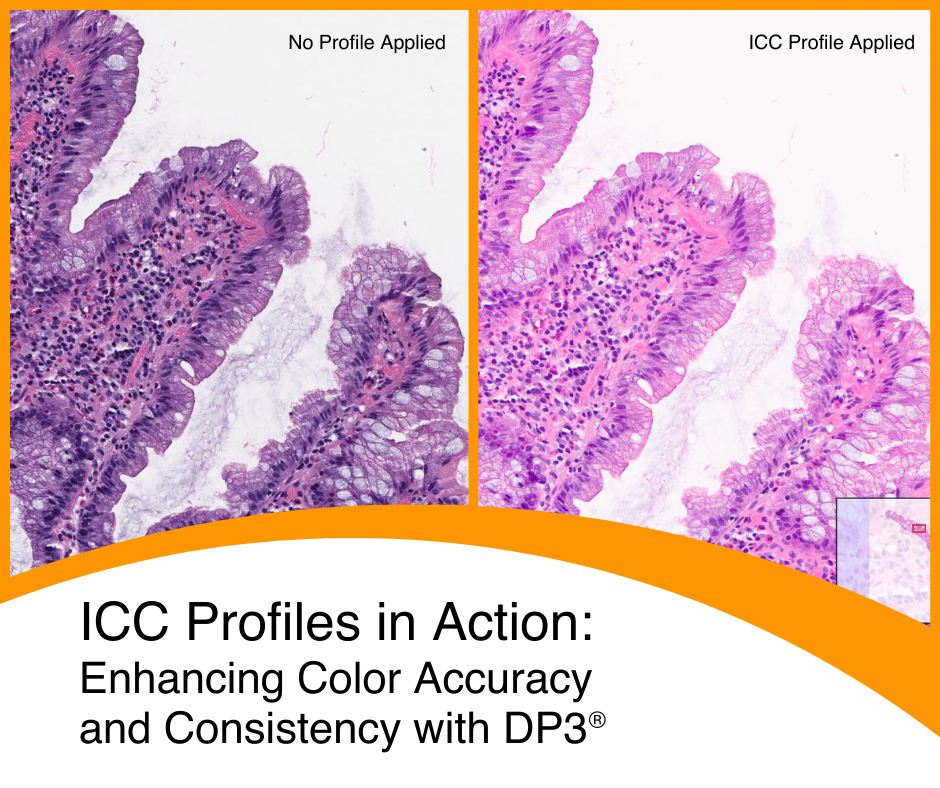
International Color Consortium: Enhancing Color Accuracy and Consistency ...
Read Now
Pathology in the Remote Work Era: Challenges and Opportunities
Read Now
There’s Always a Bigger Fish, Even in Pathology
Read Now
Digital Pathology Communication without the Keyboard – Real-time Voice ...
Read Now
Slideless Pathology: A New Era for Tumor Board Presentations
Read Now
What Will Our Legacy Be?
Read Now
Image Management in Research: Use Cases and Workflow Solutions
Read Now
Hunting for a Pathologist
Read Now
First
Prev
1
2
3
4
5
Next
Last


.png)